這個解決方案是針對有支援GPU加速的科學運算軟體所設立(如 : VASP、Molpro等 ...),我們能提供給使用者一個含有nvidia GPU的科學計算環境,其中GPU又以kepler架構為主且支援 single-precision、double precision記憶體大小於12Gb~24Gb之間,系統方面我們能做到讓使用者開機就能計算不需要煩惱過多的設定。
VASP 的運行速度現已提高2.5到4倍
維也納大學 VASP 推出支援 GPU 加速器的新版 VASP v 5.4.1,以加快研發新事物的腳步,在GPU 加速運算的加持下,研究人員完成模擬作業的時間可以加快2.5到4倍。
Several core algorithms of VASP have been ported to run on GPU accelerated hardware, e.g.:
- Blocked-Davidson and RMM-DIIS
- Hybrid functionals
- Application of the real-space projection operators
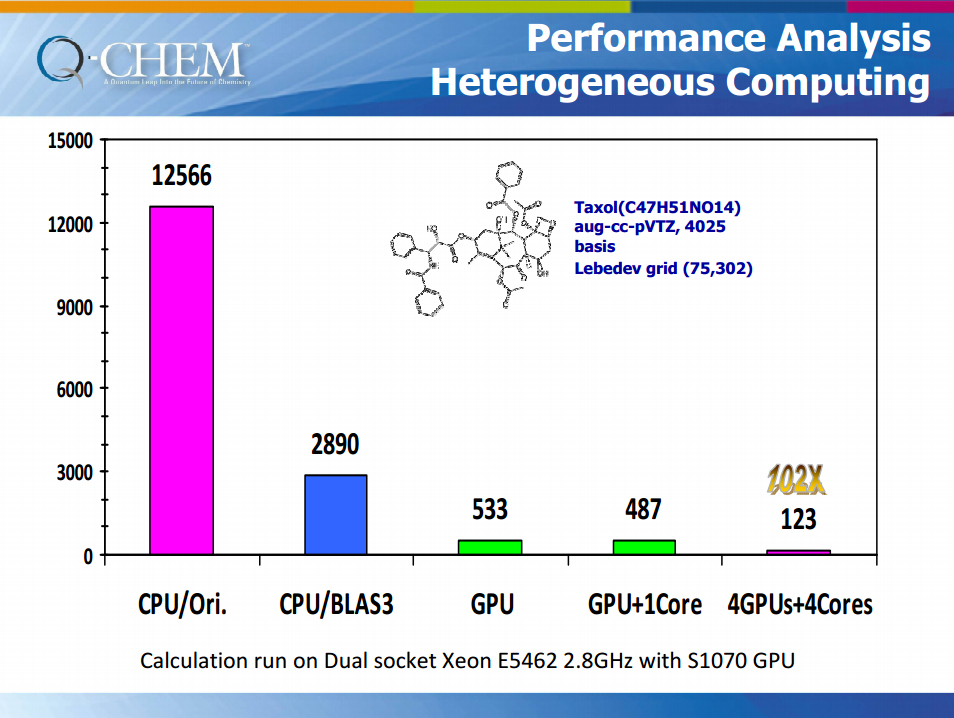
Q-Chem 的 GPU 運算效能
右圖為使用S1070 GPU及intel Xeon E5462 2.8GHz CPU 運行同一範例計算的效能差別
Q-Chem 目前提供可使用GPUs加速運算 RI-MP2 calculations .

最新版AMBER 16 GPU 全面支援 PMEMD算法,其內容包含AMBER 14 的特色重點以及新增以下特點 :
- Support for semi-isotropic pressure scaling.
- Support for the Charmm VDW force switch.
- Enhances NMR restraint support and R^6 averaging support
- Gaussian accelerated molecular dynamics.
- Expanded umbrella sampling support.
- Constant pH and REMD Constant pH support.
- Support and significant performance improvements for the latest Maxwell and the soon to be released Pascal GPUs from NVIDIA.
- Adaptively biased MD (coming soon via automatic update).
- Thermodynamic Integration, FEP and MBAR (coming soon via an automatic update).
Gaussian 16 GPU 運算加速
在Linux的作業系統環境下Gaussian 16目前能夠使用Nvidia K40和K80的GPUs來進行運算。
較早期的GPU則未有足夠的運算能力及記憶體滿足執行Gaussian 16的運算 ,且Gaussian 16也尚未支援 Tesla-Pascal 系列的GPU。
GPUs 在大型分子的 DFT energies, gradients and frequencies (for both ground and excited states) 的運算都是有效的, 但是對於較小型分子運算則是無效的 . 另外也包括 post-SCF calculations 諸如 MP2 or CCSD也是無效的.

Molpro 的 GPU運算加速
Molpro 的 "Density-fitted MP2 (DF-MP2), density
fitted local correlation methods (DF-RHF, DF-KS), DFT"算法能夠支援GPU加速但不包含 (EOM-)CCSD.
您可以聯絡我們進一步瞭解更多資訊