{GV6更新重點}{觀察分子結構及反應} { 加強圖形化功能開展化學視算技術} { 大尺寸分子的建構與管理 } {建構分子更順手} {準備 G16 的計算 }
█ GV6 更新重點 :
- Gauss View 6 可顯示IR, Raman, VCD 和 ROA光譜的諧波及非諧波頻率的分析結果,可以分別或在同一圖上以圖形方式觀察光譜。
- Gauss View 6 可顯示SCRF算法的溶劑空腔結果,且可自訂顯示為固態、網格或分子節點的介面。
- GaussView 6 現在可以繪製旋光色散排量(Optical Rotary Displacement)算法的結果。可以在單個圖面裏顯示分子組中每個分子的ORD結果。
- GaussView 6 可顯示包含電子振動頻譜和達斯欽斯基矩陣的電子震動分析結果。
- 改善 Calculation Summary
- 於Overview頁籤中新增溶劑模型和極化率的項目
- 於thermo頁籤顯示頻率,列出介由計算預測出來的熱化學量。
- 於opt頁籤顯示結構優化項目,列出當前優化步驟的收斂進度其中包括最大力值和RMS force及位移,還包括預測能量的變化。
- Gauss View 6 可將標準模式和標準震動模式的動畫儲存成MP4格式的影片檔,在儲存標準模式的動畫中無論是在正反向播放,使用者都可以設定動畫重複的頻率且可調整動畫的播放速度及流動性。振動模式動畫則提供設定位移大小的選項,以及顯示位移和 dipole derivatives的向量。
- Gauss View 6可以將多個計算結果資料來源同時顯示在一個統計圖上,且使用者可自訂下列選項
- 可改變統計圖上線的表現 : 可顯示為線型和棒型又或是兩者組合。
- 可設定線/棒特性,如厚度和顏色。
- 可繪製組合峰並設定外觀。
- 可分配資料集成分的權重包含Boltzmann averaging。
- 在Gauss View 6裏透過幾個步驟的設定即可執行一個分子群組的gaussian job,參考以下說明 :
- 確保所有分子都在一個分子組內。
- 在Gaussian Calculation Setup中設定好job類型、化學模型、標題、及其他的keywords。
- 點擊 Assign to Molecule Group 按鈕設定所有分子皆在一個群組中。
- 當儲存時設定 Save Molecule Group作用為建立每個分子的separate file,這邊可將每個分子的前綴和分子編編號添加設定至每一個分子。
- Gauss view 6使用GMMX附加模組進行構形的confrontational search,使用者可使用Cartesian search或是Bonds method的搜尋方法進行搜尋設定。
- SC Job Manager是一個於本機端Gauss View 6內建的排程系統,jobs設置好添加至排程後不需其餘步驟設定就可以自動運行。
- 對稱 - GaussView’s point-group symmetry feature allows you to increase the symmetry of the current molecule. A molecule can be constrained to a specific point group, and all future modifications to the structure will maintain that symmetry.GaussView 6 now allows you to reduce the symmetry of a molecule you are working on. Preserving some molecular symmetry is useful for setting up calculations that involve Jahn-Teller distortions. The graphic below shows the current point group being modified from D3h to D3.
- Brush Selection Tool(筆刷選取工具) - GaussView6目前提供三種不同的工具,以便在分子中選取原子。最新的添加的是筆刷工具。該工具用於快速選取密集簇中的原子。
- 最新的job設定特色
- 在Gaussian Calculation Setup中的 Job Preview 介面顯示生成的輸入檔案
- New controls enable retrieving data from checkpoint files and specifying the %OldChk directive.
- 可設定job所使用的最大硬碟空間
- New job types 新增預測 polarizabilities and ORD.
- There is an expanded panel for specifying population analysis
- 支援Gaussian 16 新版的特性功能: e.g., anharmonic IR/Raman, VCD annd ROA calculations.
- 瀏覽Default.Route檔案的內容
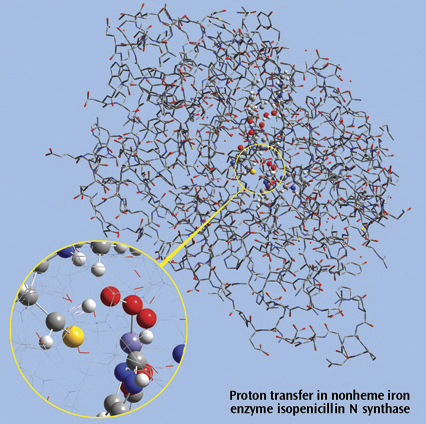
近觀左圖異青黴素N合成酵素(isopenicillin N synthase,IPNS)搭配IRC(Intrinsic reaction coordinate following) 反應路徑追蹤法的質子轉換。採用Gaussian的ONIOM法研討此5368個原子的系統,並在GaussView 內檢視。
為清楚表示分子結構, 省略部分較低層的氫原子之圖示,僅採用全分子架構及局部放大方式檢視較高的OMINON精確層。較低準確層於近視圖部分採線型框架 顯示方式,全分子部分,則以管狀方式表示。參考M. Lundberg, T. Kawatsu, T. Vreven, M. J. Frisch and K. Morokuma, JCTC 5 (2009) 222。
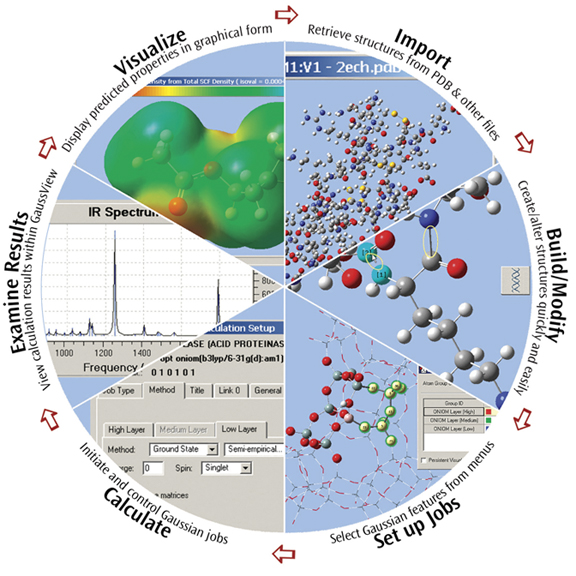
GaussView提供 Gaussian最先進、功能強大的圖形界面。有了GaussView就可輸入及建構想要的分子結構, 從 "設定"、"啟動"、"控制並檢視Gaussian計算",接著取得並檢查結果,都不需要離開GaussView軟體。
GaussView 6(GV6)提供化學研究人員方便直接方法處理大尺寸的分子系統,GV6也完全支援G0 新的功能及分子建構方式。
█ 大尺寸分子的建構與管理
GV6提供研究大尺寸分子系統,從PDB檔,G09每一階段該有的功能都可圖形化處理,開始讀入分子資料,改變分子結構與設定ONIOM計算。 最後再檢視及繪製計算結果。此外GV6亦可讀取許多常用的資料輸入格式。
GV6完全支援從PDB檔讀入的分子結構,並直接進入計算工作:
- 自多結構文件選擇所需的結構。
- 依據使用者習慣選擇自動或手動,在所有原子上添加氫原子。
- 選擇性添加氫原子到單一或多個副產物、化學鏈、螺旋或其他已定義的實體結構。
- 在個別副產物或次結構標出或選擇原子。
- 利用滑鼠快速標出原子副產物之成分。
- 依據不同的彈性準則指定給 ONIOM 層。
- 保留G16計算中的殘基訊息,並取得G16計算結果。
GV6 提供較 GaussView 早期版本更彈性的功能:
- GV6提供一組常見的工具盤視窗,若需要一組平面八碳環類的素材,則可透過顯示的建構工具視窗從自訂建好的分子結構資料庫擷取。
- 從元素週期表版面選取 X原子符號的啞原子。
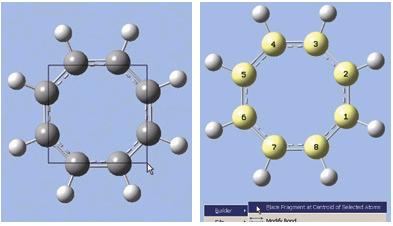
- 現在選用所有需要的碳原子。方法之一是採用GV5新的rubberband 選擇模式。在視窗上,按住R鍵並拖曳滑鼠,框出一個接觸到所有碳原子的矩形。當鬆掉滑鼠按鍵時,被選到的碳原子會呈現黃色
- 在視窗上單擊右鍵,然後移至選到的原子再從內容選單選擇 Builder→Place Fragment。此時環的中心就出現一個紫紅色的的啞原子。
- 按下Rebond游標;把分子鍵從啞原子加到選到的八個碳原子。
- 點擊Add Valence添加原子價按鈕,然後移到啞原子。 將出現一個氫原子結合到垂直於平面環的啞原子。更改氫原子為鈾原子。出現的分子如右圖。
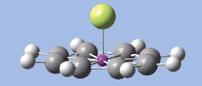
- GV6內選擇整個分子的快速方法是在視窗打開時按A鍵。 然後鍵入Ctrl-C或輸入命令-C,將分子複製到剪貼板。 剪貼板上的任何分子結構都會出現在自訂的元件選單, 選擇剪貼板,點選適當位置,加入另一單元於視窗內。
- 按住Alt鍵,限制鼠標對新組件的操作, 並將其移到所需位置附近。
- 新增一組長2.24A三價鍵 然後調整 XUUX 二面角為180°。
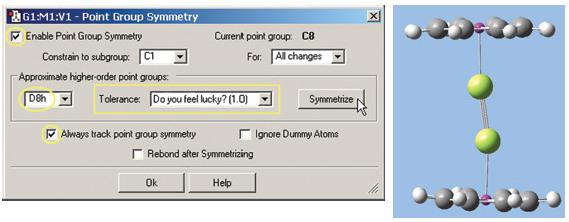
- 最後透過Point Group Symmetrg對談視窗(Edit視窗內)加強分子的對稱性。
GV6 透過圖形化機制,設定特定計算型態,全力支援G16所有功能:
- 多種方法指定原子於ONIOM層。GV6也簡化指定分子力學原子的類型與電荷。
- CASSCF及其他工作對MOs的重排與重配。
- 增加及重訂重覆的內部座標。
- 幾何優化過程中指定鎖定的原子及座標。
- 設定STQN等價原子激態最佳化。
- 常態分析(頻率計算)選定原子。
- 指定核磁共振自旋耦合的原子。
- 建構用於週期邊界條件(PBC)計算的聚合物單位細胞、二維表面和晶體。